
手机扫码访问本站

微信咨询
衣康酸(itaconate)是免疫过程中代谢重编后的最好例子之一。它是由三羧酸循环(TCA循环)中的顺势乌头酸(cis-aconitate)在巨噬细胞中合成的,巨噬细胞被多种因素激活,尤其是脂多糖(LPS),以及其他Toll样受体(TLR)配体和细胞因子,如I型和II型干扰素[1-4],这些刺激增加了乌头酸脱羧酶1 (ACOD1;也被称为CAD),最初叫免疫响应基因1蛋白质(IRG1),顺式乌头酸从TCA循环中转移,重新调整其用途以产生衣康酸(图1)。目前的研究表明,衣康酸是一种重要的免疫代谢产物,对免疫、宿主防御和肿瘤发生具有深远的影响。
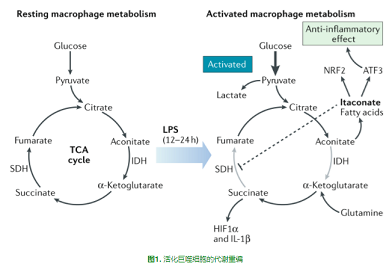
IRG1已被证明定位于线粒体[5],这是巨噬细胞活化过程中活跃的代谢重构位点。在促炎触发后,巨噬细胞向糖酵解表型转变,而线粒体功能则随着耗氧量的降低而下降[6]。线粒体功能障碍是TCA循环中两个断点的结果,这两个断点使线粒体功能丧失[7]。突变点与琥珀酸(succinate)的积累[8]和通过异柠檬酸(isocitrate)脱氢酶阻断代谢通量有关[7]。通过琥珀酸脱氢合酶(SDH)进行的琥珀酸氧化可以在复合物I处进行反向电子输运,从而驱动活性氧物质(ROS)的生成以促进缺氧诱导因子1α(HIF1α)和IL-1β的生成[8]。IRG1介导的衣康酸积累在巨噬细胞的促炎性激活过程中引起琥珀酸积累[9-10]。衣康酸能直接抑制来源于原代巨噬细胞的SDH的酶活性。衣康酸直接抑制SDH的能力来源于其与琥珀酸的结构相似性(图2),衣康酸和经典的SDH抑制剂丙二酸(malonate),可以竞争性地阻断SDH活性位点[11]。
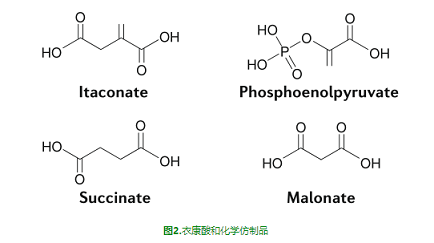
衣康酸盐抑制SDH的能力是衣康酸盐的一个显著的代谢调节特征,表明衣康酸盐在缺乏IRG1的巨噬细胞中的免疫调节作用可能部分归因于这种抑制作用。
NRF 2最初被认为是氧化应激的传感器,它被一种叫做Kelch-like ECH相关蛋白1(Keap1)的蛋白质所控制,这种蛋白质以NRF 2为靶点,用于蛋白质体降解[12]。然而,在氧化应激反应中,KEAP1被灭活,NRF2被释放,诱导大量NRF2依赖基因的转录。由此产生的基因产物在很大程度上保护了应激诱导的细胞死亡,特别是氧化应激的细胞毒性作用。NRF2被认为是感染和炎症过程中损伤的关键控制因子[13]。这种控制的发生是因为它能够增加编码酶的基因的表达,如血红素加氧酶1和参与合成谷胱甘肽(glutathione)的酶,谷胱甘肽是防止细胞氧化应激的主要保护剂。
这些发现使NRF2成为一种新的抗炎方法的有吸引力的靶点。衣康酸的两个衍生物,2-甲基衣康酸和4-辛基衣康酸均具有细胞渗透性,能激活NRF2并在巨噬细胞中驱动NRF2依赖性基因表达[14-15]。衣康酸激活NRF2除了诱导NRF2依赖基因外,还会产生下游效应。衣康酸抑制IL-1β诱导的能力包括抑制SDH(如上所述),但也在一定程度上涉及Nrf2的激活[15]。这可能是由于NRF2直接抑制IL1B基因转录,也可能是通过诱导抗氧化剂(如谷胱甘肽)产生的,谷胱甘肽会限制ROS,这已被证明是诱导IL-1β所必需的[16]。
因此,总的来说,衣康酸对NRF2的作用将通过诱导抗炎基因(如编码血氧合酶1的基因)来控制炎症,这些基因通过升高的谷胱甘肽来限制ROS的炎症作用,也可能直接阻断促炎基因的转录。图3描述了衣康酸作为半胱氨酸(cysteine)修饰剂在靶蛋白上的作用,这些修饰可能导致免疫功能的改变。
图3. 衣康酸可修饰蛋白质以引起抗炎作用.
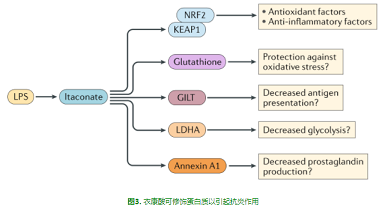
作为一种α,β-不饱和羧酸,衣康酸有一个经典的亲电骨架,允许它接受来自被称为亲核细胞的潜在相互作用伙伴的电子对。在细胞环境中,亲电化合物通常以含有巯基(–SH)基团的蛋白质为目标,从而诱导亲电应激反应(ESR)。谷胱甘肽是细胞中最普遍存在的分子之一,它通过中和细胞中多余的亲电体,然后分泌谷胱甘肽亲电加合物,提供了对抗亲电应激的第一线防御。例如,直接质谱测定表明,内源性衣康酸与谷胱甘肽形成加合物[14]。ESR通常由KEAP1通过其反应性半胱氨酸残基检测到,使其类似于氧化应激反应。虽然KEAP1-NRF2被认为是ESRs的主要调控因子,但几个KEAP1独立或NRF2独立的通路也被亲电应激触发:热休克反应、自噬溶酶体通路和内质网应激反应[17]。这些途径受许多调节因子控制,包括ATF3、AF4、p62和热休克因子蛋白1[17]。因此,ESR是由这些途径的激活程度来定义的,几乎每个亲电分子在激活ESR成分时都有一个独特的足迹。
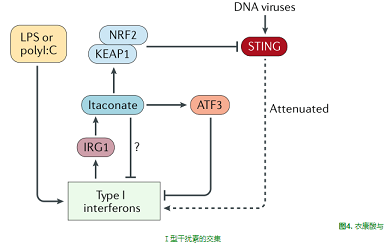
衣康酸及其衍生物为研究ESR谱提供了独特的机会。在缺乏IRG1的巨噬细胞中的实验表明,内源性衣康酸除了NRF2激活外,还可以诱导ATf3蛋白和ATf3驱动的应激反应[14]。ATF3是一种免疫激活的全球性负调节因子,可以调节IL-6等细胞因子[18],也是与线粒体应激相关的主要调节因子[19],在代谢紊乱和炎症信号之间提供了额外的联系。
最近研究表明,NRF2可抑制干扰素基因(STING)的刺激因子,后者是病毒感染期间I型干扰素产生的关键驱动因素,4-辛基-衣康酸可抑制STING,并抑制Ⅰ型干扰素在对STING活化剂和病毒的应答中的表达[20]。4-辛基-衣康酸的作用依赖于NRF2,因为4-辛基-衣康酸不能阻断NRF2缺陷细胞中的STING。4-辛基-衣康酸还抑制了婴儿期发病的被称为STING相关血管病变的患者的成纤维细胞产生Ⅰ型干扰素,这些患者在编码STING的基因第5外显子中有功能点突变。4-辛基-衣康酸也被证明能有效地阻断脂多糖或聚肌苷-聚胞苷酸(polyⅠ:C)诱导的Ⅰ型干扰素[15],进一步表明Ⅰ型干扰素的产生可能是衣康酸一个特别重要的靶点。内源性衣康酸参与调节干扰素信号的作用和确切机制尚待确定。衣康酸与I型干扰素的交叉点如图4所示。
防止炎症巨噬细胞(也称为M2巨噬细胞)分化将增加IRG1的表达和衣康酸的产生[21-22]。过氧化物酶体增殖物激活受体γ(PPARγ)是脂肪生成和脂肪细胞功能的主要调节因子,已被证实参与了M2巨噬细胞的激活。PPARγ被证明是IL-4依赖基因表达所必需的,PPARγ缺乏导致IRG1表达增强[21]。有可能,去除炎症巨噬细胞诱导的制动器(PPARγ形式)导致衣康酸的补偿性增加,以恢复体内平衡。图5a所示的IRG1和衣康酸在M2巨噬细胞极化中的可能作用。
衣康酸在另一个巨噬细胞群——肿瘤相关巨噬细胞中的作用,被认为与M2巨噬细胞有点类似[23]。在涉及B16黑色素瘤细胞或ID8卵巢癌细胞的腹膜肿瘤中,一种无偏倚的代谢组学筛选鉴定出,衣康酸是腹膜组织驻留巨噬细胞中高度上调的代谢产物之一。IRG1基因的敲除显著减少了腹腔肿瘤的发生,表明衣康酸具有促肿瘤作用。其机制似乎与衣康酸促进氧化磷酸化有关,而氧化磷酸化反过来又会增加ROS的产生,从而增加肿瘤的生长。图5b显示了肿瘤相关巨噬细胞产生的衣康酸作为肿瘤前代谢物的可能作用。
衣康酸可以驱动人类单核细胞的先天免疫耐受(即对炎症刺激的反应降低)[24]。β-葡聚糖是一种真菌细胞壁成分,已知能长期上调单核细胞固有免疫功能,通过降低IRG1的表达来抵消耐受性。β-葡聚糖也增加了SDH的表达,促进氧化磷酸化。重要的是,编码IRG1和SDH的基因多态性被描述为调节耐受性的诱导。衣康酸在巨噬细胞免疫麻痹中的作用如图5c所示。
对于一种在19世纪首次被描述,然后在哺乳动物系统中被忽视的代谢物,过去几年里,人们对衣康酸的兴趣有了显著的提高。对衣康酸免疫作用的研究已经表明了治疗多种疾病的可能性。以SDH为中心和亲电方式的衣康酸作用均表明衣康酸具有明显的抗炎活性。衣康酸或其衍生物修饰多种炎症蛋白,包括NRF2和ATF3,表明衣康酸在多种疾病中可能具有治疗潜力。其与肿瘤生长、M2巨噬细胞功能、免疫麻痹的联系,都为进一步分析衣康酸在健康和疾病中的作用提供了诱人的前景。
中科脂典(中科院遗传发育所参股孵化)是一家主要从事前沿生物技术开发、生物技术服务、生物制品研发及销售的高科技公司。公司在脂组学、代谢组学、蛋白组学等领域开发了具有国际前沿水平的组学技术。公司提供基于液相色谱连接质谱的靶向代谢组学检测服务,欢迎有衣康酸、TCA循环等检测需求的客户咨询。
[1]. Shin, J. H. et al. (1)H NMR- based metabolomic profiling in mice infected with Mycobacterium tuberculosis. J. Proteome Res. 10, 2238–2247 (2011).
[2]. Strelko, C. L. et al. Itaconic acid is a mammalian metabolite induced during macrophage activation. J. Am. Chem. Soc. 133, 16386–16389 (2011).
[3]. Sugimoto, M. et al. Non- targeted metabolite profiling in activated macrophage secretion. Metabolomics 8, 624–633 (2012).
[4]. Michelucci, A. et al. Immune- responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl Acad. Sci. USA 110, 7820–7825 (2013).
[5]. Degrandi, D., Hoffmann, R., Beuter- Gunia, C. & Pfeffer, K. The proinflammatory cytokine- induced IRG1 protein associates with mitochondria. J. Interferon Cytokine Res. 29, 55–67 (2009).
[6]. Krawczyk, C. M. et al. Toll- like receptor- induced changes in glycolyti metabolism regulate dendritic cell activation. Blood 115, 4742–4749 (2010).
[7]. Jha, A. K. et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42 419–430 (2015).
[8]. Tannahill, G. M. et al. Succinate is an inflammatory signal that induces IL-1bet through HIF-1alpha. Nature 496, 238–242 (2013).
[9]. Cordes, T. et al. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J. Biol. Chem. 291, 14274–14284 (2016).
[10]. Lampropoulou, V. et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 24, 158–166 (2016).
[11]. Ackermann, W. W. & Potter, V. R. Enzyme inhibition in relation to chemotherapy. Proc. Soc. Exp. Biol. Med. 72, 1–9 (1949).
[12]. Ahmed, S. M., Luo, L., Namani, A., Wang, X. J. & Tang, X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 585–597 (2017).
[13]. Hayes, J. D. & Dinkova- Kostova, A. T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 39, 199–218 (2014).
[14]. Bambouskova, M. et al. Electrophilic properties of itaconate and derivatives regulate the IkappaBzeta- ATF3 inflammatory axis. Nature 556, 501–504 (2018).
[15]. Mills, E. L. et al. Itaconate is an anti- inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117 (2018).
[16]. Mills, E. L. et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167, 457–470 (2016).
[17]. Levonen, A. L., Hill, B. G., Kansanen, E., Zhang, J. & Darley- Usmar, V. M. Redox regulation of antioxidants, autophagy, and the response to stress: implications for electrophile therapeutics. Free Radic. Biol. Med. 71, 196–20 (2014).
[18]. Gilchrist, M. et al. Systems biology approaches identify ATF3 as a negative regulator of Toll- like receptor 4. Nature 441, 173–178 (2006).
[19]. Quiros, P. M., Mottis, A. & Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 17, 213–226 (2016).
[20]. Olagnier, D. et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat. Commun. 9, 3506 (2018).
[21]. Nelson, V. L. et al. PPARgamma is a nexus controlling alternative activation of macrophages via glutamine metabolism. Genes Dev. 32, 1035–1044 (2018).
[22]. Ganta, V. C. et al. A microRNA93-interferon regulatory factor-immunoresponsive gene-1-itaconic acid pathway modulates M2-like macrophage polarization to revascularize ischemic muscle. Circulation 135, 2403–2425 (2017).
[23]. Weiss, J. M. et al. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J. Clin. Invest. 128, 3794–3805 (2018).
[24]. Dominguez- Andres, J. et al. The itaconate pathway is a central regulatory node linking innate immune tolerance and trained immunity. Cell Metab. 29, 211–220 (2018).