
手机扫码访问本站

微信咨询
舒尼替尼 ( Sunitinib、SU011248) 是一种多靶点的生物靶向抗肿瘤药物,能同时抑制多条信号传导通路,具有抗肿瘤和抗血管生成作用。舒尼替尼在肾细胞癌 (RCC )和胃肠 间质瘤 (GIST ) 的临床前和临床研究中均显示出了确切的抗肿瘤效应。2006年 1 月美国食品和药品管理局 ( FDA ) 正式批准了舒尼替尼用于治疗上述两种肿瘤。此外,舒尼替尼在其他实体瘤的研究中也显示出了一定的作用。
舒尼替尼是一种口服的多靶点的酪氨酸激酶抑制剂。通过抑制 VEGFR、PDG FR、Kit 和 RET等酪氨酸激酶而阻断信号传导通路,最终影响恶性肿瘤的生长、增殖和转移。在晚期 RCC和 IM治疗失败的 GIST患者中舒尼替尼表现出了确切的抗肿瘤效应,美国 FDA已批准舒尼替尼应用于上述 2种肿瘤。此外,在神经内分泌肿瘤、NSCLC、结肠癌、原发性肝癌和乳腺癌的临床研究中舒尼替尼也显示出了活性。
目前,舒尼替尼单药治疗的研究在多种的恶性肿瘤中展开,而联合其他药物的临床研究还处于初始阶段,关于联合应用的耐受性及最佳的治疗方案仍有待进一步探讨。随着临床研究的深入。
片剂:12.5 mg/片,25 mg/片,50 mg/片。
适用于胃肠间质瘤对伊马替尼耐药或者治疗后进展者,或用于肾细胞癌的治疗。
适用于胃肠间质瘤对伊马替尼耐药或者治疗后进展者,或用于肾细胞癌 的治疗。在一项随机、双盲、安慰剂对照的国际临床实验中,312例使用甲磺酸伊马替尼后不耐受或者病情恶化的 G IST患者,按照 2∶1比例被随机分入给药组和安慰剂对照组。给药组口服 50 m g舒尼替尼,1日 1次。一个疗程持续 4周,停药2周后继续用药,直到患者病情恶化,或者由于其他原因出组。结果表明,用药组患者的肿瘤进展中位时间 (TTP)为 27. 3周,安慰剂组为 6.4 周 (P <
0. 000 1)。
给药组的无进展生存中位时间 (PFS )为 24.1 周,安慰剂组为 6.0 周。在另外一项多中心、开放试验 Ⅱ中,55 名患者中,有 9.1%患者的肿瘤得到部分缓解(partial response,PR)。晚期肾细胞癌适应证的批准主要是基于部分缓解率(partial response rate)和缓解时间 (duration of response)而定的。两项多中心、开放、无对照试验结果表明,对于细胞因子生物治疗无效的晚期肾细胞癌患者,使用本品的客观缓解率(objective response rate,ORR)分别为 25. 5%和 36.5%:缓解时间分别达到 27.1。
舒尼替尼是一种抑制多种受体酪氨酸激酶的小分子化合物,而一些酪氨酸激酶参与了恶性肿瘤的生成、病理性血管生成以及肿瘤转移扩散的过程。生化、细胞学实验证明,舒尼替尼能够抑制 80多种酪氨酸激酶,和多种生长因子受体,如血小板源性生长因子受体(platelet- derived grow th factor receptors,PDGFR)、血管内皮生长因子受体 (vascu lar endothelial grow th factor receptors,VEGFR)、Ⅰ- 型集落刺激因子受体(colony sti m ulating factor receptor type 1,CSF- 1)、细胞因子受体 (stem cell feztor receptor,K IT)等。
在表达酪氨酸激酶的肿瘤表达体系中,舒尼替尼能够抑制多种酪氨酸激酶的磷酸化过程。通过实验性肿瘤模型证实,舒尼替尼有抑制肿瘤细胞生长、退化、转移的作用。对于酪氨酸激酶调控失灵肿瘤细胞, 舒尼替尼能够抑制其生长。
药物代谢动力学数据主要来自于健康志愿者和一些晚期的癌症患者,其中还 包括 RCC和 GIST患者的临床研究。在晚期肿瘤患者中,舒尼替尼 50mg /d连续口服 28天。
第 1天,舒尼替尼及其代谢产物 SU012662的平均最大血药浓度 (Cmax) 分别为 27.7n g /m l 和 4.12ng /ml,中位达峰时间约 5h。0~24h的平 均血药 浓度 -时间曲线下面积(AUC0~24 ) 分别为 420 ng·h/ml 和 63.6ng·h /ml。第 28天平均 C max 分别为 72.2ng /ml 和 33. 7ng /ml,达峰时间分别为8.5 h和6.5h,AUC 0~24分别为 1 296 ng·h/ml 和 592 ng·h/ml。
舒尼替尼及 SU012662的血浆蛋白结合率分别为 95%和90%,舒尼替尼的表观分布容积大约为2 230L,肌酐清除率34~64L /h,平均消除半衰期 ( t1 /2 ) 为 50h,代谢产物的 t 1 /2为95h。食物不影响舒尼替尼的生物利用率和活性代谢产物暴露率。舒尼替尼在健康志愿者和肿瘤患者的药代动力学特征无明显差异。
舒尼替尼主要通过 P450-3A4(CYP3A4 ) 代谢,产生的活性代谢产物 SU012662占总暴露率的 23%~27 %。给药剂量的 61%以原形药经粪便排出,16 %通过肾清除。舒尼替尼的药代动力学不受肿瘤类型、年龄、性别或体重影响。
常见骨髓抑制和淋巴细胞减少、腹泻,恶心,呕吐,消化不良,厌食和腹痛、乏力、口腔炎和味觉改变、皮肤脱色、高血压。重度骨髓抑制和静脉血栓栓塞、脱发不常见,偶见皮疹和手足综合征、出血、虚弱、头痛、关节痛、肌痛、口腔痛和背痛、咳嗽和呼吸困难、充血性心力衰竭、肝酶和胰酶升高及其他肝功能异常、轻度肾功能异常和电解质紊乱、甲状腺功能低下、水肿。罕见致命性胃肠道并发症包括穿孔。可能导致肾上腺功能不全。
若出现充血性心力衰竭的症状应停药。射血分数<50% 以及射血分数低于基线20%的患者也应停药或减量。本品可延长QT 间期,且呈剂量依赖性。应慎用于有QT间期延长病史的患者、服用抗心律失常药物、有心脏疾病和电解质紊乱的患者。若出现严重高血压 应暂停使用,直至血压恢复正常。不良反应包括疲乏、皮肤毒性(皮疹、瘙痒、脱屑和手足综合征)以及消化道反应(恶心、呕吐、腹泻和食欲缺乏)。育龄期妇女接受治疗时应避孕。哺乳期妇女应停止哺乳。
1. 圣约翰草 (S t.John ’ sW ort )可能降低本品血药浓度,服药期间不宜同服。
2. 与 CYP3A4强效抑制剂合用时可减量至 37.5 m g。与利福平等 CYP3A4 诱导剂合用时剂量最大可增至 87.5 mg,但需密切观察临床表现。当与酮康唑等强的 CYP3A4抑制剂联用时,舒尼替尼的ρm ax和 AUC0- ∞会提高 50%左右。当与利福平等强效 CYP3A4诱导剂合用时,舒尼替尼的 ρmax降低 23%,AUC0- ∞降低 46%。如果必须联合使用,需适当调整给药剂量。
3. 舒尼替尼对于细胞色素 P450酶系没有诱导或者抑制作用。
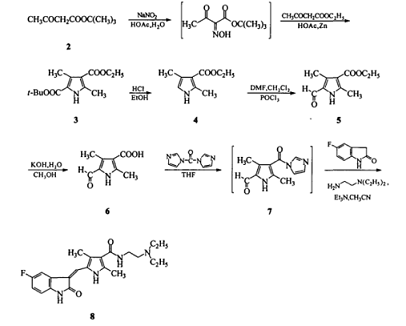
以乙酰乙酸叔丁酯为起始原料,通过 Knorr 吡咯合成法得到3,5 -二甲基-1H-吡咯-2,4-二羧酸-2-叔丁酯-4-乙酯( 3),然后经 5 位脱叔丁氧羰基、Vilsmeier 甲酰化、酯水解反应得到关键中间体 5-甲酰基-2,4-二甲基-1H-吡咯-3-羧酸( 6),采用“ 一锅煮”方法,化合物 6 与碳酰二咪唑( CDI)反应生成酰基咪唑( 7) 后不经分离直接与 5 -氟吲哚啉-2-酮和 2- (二乙氨基) 乙二胺反应得到舒尼替尼:
[1] 陈逢生, 石敏, 罗荣城. 多靶点酪氨酸激酶抑制剂舒尼替尼的研究进展[J]. 临床肿瘤学杂志, 2008, 13(3): 278-281.
[2] 郭婕, 罗鹃, 朱珠. 抗肿瘤新药——舒尼替尼[J]. 中国药学杂志, 2007, 42(13): 1037-1038.
[3] 常用新药精汇手册
[4] 新编妇幼专科用药速查手册
[5] 董肖椿, 周福生, 闻韧. 抗肿瘤药物苹果酸舒尼替尼的合成[D]. , 2008.