
手机扫码访问本站

微信咨询
利奈唑胺合成路线至少七步,使用了保护基团。
Timothy F. Jamison等设计了会聚式路线,不需要保护基。
逆合成分析路线如图1所示:
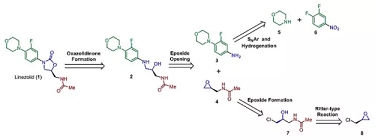
图1. 利奈唑胺的逆合成分析
从制备酰胺7开始,(+)-环氧氯丙烷8和乙腈发生Ritter型反应。用三氟化硼乙醚作为化学计量的Lewis酸,室温下反应5 min,反应结束用NaHCO3溶液淬灭,间歇式反应纯化得到7,平均收率62%。
副产物9的生成很大程度上被抑制,却生成了约15%的副产物10,乙醚作为亲核试剂参与了反应。
对此,作者更换了位阻更大的三氟化硼丁醚,消除了副产物,同时收率提高至90%,图2所示。
7的反应较为理想了,还有就是提纯,作者尝试使用T-型混合装置将水溶液淬灭步骤整合到连续流动装置体系中,但反应后的硼酸析出沉淀容易堵塞通道,换为异丙醇在-35 ℃下淬灭Lewis酸后生成了可溶性的硼酸酯副产物。
在这个反应条件下,中间体腈鎓离子也会和异丙醇反应,生成亚氨酸酯12,并通过核磁氢谱得以验证。随后将12的流动相通入含有1:1比例的THF和1,2-二氯乙烷混合溶剂中,以叔丁醇锂为碱,反应温度-35 ℃,停留反应时间4 min,得到环氧化物13。
其中1,2-二氯乙烷能够在低温下溶解反应生成的氯化锂。这样以(+)-环氧氯丙烷8为起始物料,反应时间仅为10.2 min,得到一个酰胺保护结构的环氧化合物13。
对反应器II和反应器III中的条件的变化,如异丙醇和叔丁醇锂的当量,甚至包括冷浴温度和滞留反应时间的变化,都会使氨基醇2的收率下降。
12中活泼胺基官能团的保护,也降低了在反应器III中合成环氧时的副产物的形成。
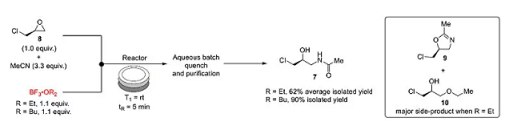
图2. 化合物7的合成
中间体苯胺3的合成都是常规反应,芳香亲核取代反应SNAr,氢化还原。
吗啉5和3,4-二氟硝基苯6在150 ℃下在1,4-二氧六环和DMF的混合溶剂中生成14,需要反应10 min。
反应混合物通入氢气,质量流量控制器控制(氢气:液体流速为15:1),控制反应温度100 ℃,背压100 psi,通过不锈钢Pd(0)填充床,滞留时间仅为0.33 min,中间体14便完全转化为3,没有脱卤副反应发生。
1,4-二氧六环和DMF混合溶剂是反应成功的关键之一,它能加速SNAr反应、溶解所有物料和副产物,与钯填充床兼容。
氢气排出后,3和13在反应器Ⅵ中生成2,反应温度40 ℃,时间12 min,而且不需要额外的活化试剂。
在氢化还原时有水生成,亚氨酸酯水解为对应的酰胺结构。
减少滞留时间或提高温度没有提升收率。快速氧化,氨基醇2和苯胺3难以从粗品反应混合物种提纯分离,使用了连续流动化学技术,这两个物料快速反应消耗,氧化损伤可以降至最低。
恶唑烷酮环的合成反应,由物料2的流动相在反应器Ⅶ中和羰基二咪唑的二氧六环溶液,150 ℃反应5 min,其他的缩合试剂如光气衍生物,因反应混合物中还有异丙醇等亲核试剂不能生成1。
反应结束后,加入HCl在线分离,离线提纯。工艺路线如图3所示:
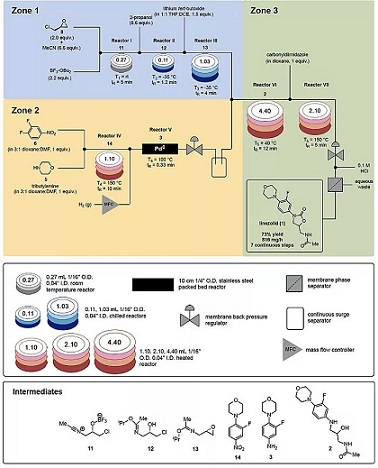
图3. 具体反应条件和中间体结构
利奈唑胺的收率73%,总时间27 min,产量816 mg h-1,液相分析验证了光学纯度在反应全程中能够保持。间歇式反应的总时间则大于60 h。
E-因子(E-factor)是评估产品制造过程中对环境影响度的参数,E = Kg废物/Kg产品,E值越大,合成同样的产品过程中产生的废物就越多。这个流动化学工艺的E-因子仅为25,平均每步3.57,而工业化药物合成工艺每步的E-因子为25-100,相比之下流动化学优势极其明显。
在这项工作里,作者的连续流动合成涉及七个不同反应步骤,而无需溶剂交换或中间纯化,这已经创下了目前的最高纪录。而且他们所用的设备也常见,在一个通风橱里即可实现(图4)。这正是化学合成工艺不断改进的方向所在,通过合理设计路线和选择物料,简洁、高效、环保、快速地得到目标产物。同时,这也体现了连续流动化学在有机合成中的优势。

图4. 工艺中所使用的合成设备
Nature Reviews Chemistry 的主编Stephen G. Davey博士还在Research Highlight栏目中撰文[1] 介绍了这种不需要中间停顿的合成工艺。
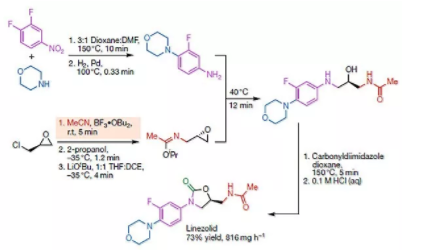
图5. 利奈唑胺的合成路线