
手机扫码访问本站

微信咨询
依泽替米贝(Ezetimibe)是由美国先灵葆雅制药公司(Schering-Plough和默克公司(Merck)联合研发的一款新型降脂药。药理作用研究表明:依泽替米贝是一种新型的选择性胆固醇吸收抑制剂,可以通过影响小肠刷状缘摄取和转运胆固醇微胶粒的载体活性,从而抑制肠壁吸收食物和胆汁中的胆固醇和植物固醇而降低体内胆固醇的水平。实验证实:依泽替米贝对小肠和肝脏胆固醇的合成没有直接影响,只能通过阻止肠壁囊泡中的外源性胆固醇转运至细胞内胆固醇池,抑制外援性胆固醇转移到淋巴,但并不影响新合成的胆固醇(内源性)合成到小肠的脂蛋白中。
临床上,依泽替米贝被用于高胆固醇血症的治疗,可以显著降低高血脂症患者的血浆低密度脂蛋白胆固醇(LDL-C)、总胆固醇(TC)和龋齿类对甘油三酯(TG)水平,并升高高密度脂蛋白胆固醇(HDL-C)水平。当与其他汀类药物联合使用时,可同时抑制胆固醇的内源和外源代谢途径,可以更好地控制高血脂症患者的血脂水平,取得显著地降低血脂和抗动脉粥样硬化效果。依泽替米贝同时也可用于两种不常见的高血脂症治疗:纯合子家族遗传性高胆固醇血症和纯合子谷固醇血症。
依泽替米贝于2002年10月经过美国FDA批准上市,商品名“益适纯(Zetia/Ezetrol)。目前已经在超过90个国家和地区上市销售。依泽替米贝在2006年经过国家食品药品监督管理局(SFDA)批准在中国获得注册销售,2007年下半年开始正式起步销售,目前已在多地纳入了医保目录中。
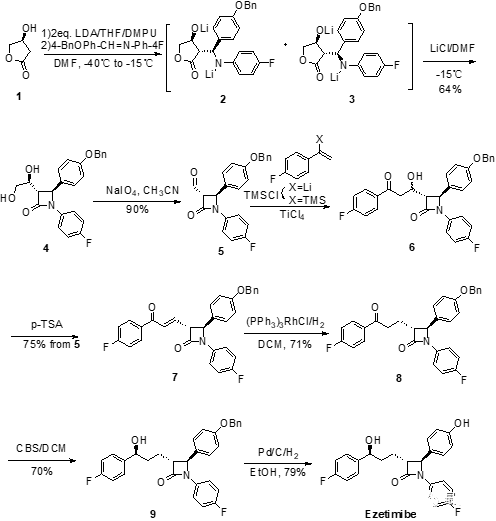
目前已经报道的用于依泽替米贝合成的方法有多种,下面我们对几种合成方法进行介绍:
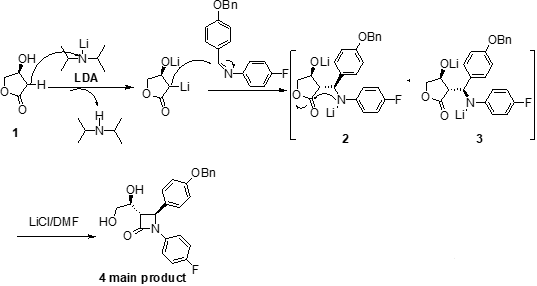
首先,依泽替米贝可以通过利用市售的(S)-3-羟基-γ-内酯1一步合成非对映选择性的反式-β-内酰胺4开始。原料1在四氢呋喃中用二异丁基胺基锂(LDA)处理后加入N-(4-苄氧基)甲苯-N-(4-氟)苯基亚胺和N,N-二甲基丙烯脲(DMPU)可得到2和3的混合物,其中2为主要产物,其比例为2:3=79:21。DMPA是一种优良的有机溶剂,毒性极低,耐光热,耐酸碱,可溶解各种有机物,无机物,也可作为高反应性亲核试剂和碱的助溶剂,使用过程中经常可代替具有致癌性的六甲基磷酰三胺(HMPA)。2和3由于形成了稳定的二价负离子锂盐聚合物而无法直接闭环得到中间体4。而当加入了N,N-二甲基甲酰胺(DMF)的氯化锂(LiCl)溶液后,锂盐聚合物发生溶解,中间体分子内发生闭环得到内酰胺主产物4(trans:cis=95:5)。可能由于中间体3胺基和酯羰基空间上距离比较远,显负电性的氮难以与羰基发生亲核反应,所以其对应的四元环内酰胺产物更加难以生成。
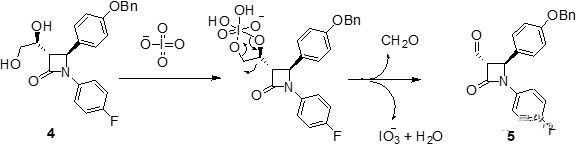
内酰胺中间体4结构中的邻二醇结构经过高碘酸钠氧化后发生碳-碳键断裂得到醛中间体5。利用相山-阿德尔缩合反应(Mukaiyama-Aldol condensation),在Lewis酸TiCl4活化作用下,4-氟苯乙烯醇的四甲基硅烷化衍生物和中间体5的醛基发生亲核加成反应得到缩合产物6。
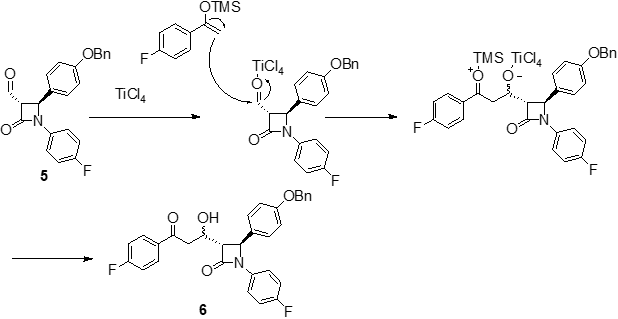
传统的Aldol缩合反应也称为醇醛缩合反应,是一个烯醇离子和羰基化合物缩合而形成一个β-羟基化合物的反应,有时可接着脱水生成共轭的烯酮产物。通常的醇醛缩合反应往往存在选择性差,副产物多的缺点,大大限制了其应用范围。1973年,日本化学家相山光昭(Teruaki Mukaiyama)对Aldol缩合反应进行了改进,将烯醇负离子转化为硅醚结构进行稳定和分离,然后再用其与醛酮进行反应制备β-羟基化合物或共轭烯酮。经过硅醚化的烯醇化合物其亲核反应性能减弱,需要在Lewis酸或碱的帮助下才能实现与醛酮的反应。不经过手性控制的条件下,相山-阿德尔醇醛缩合反应一般得到的是外消旋混合物。当然,目前也有很多文献和工作报道了对映选择性控制的不对称相山-阿德尔反应。
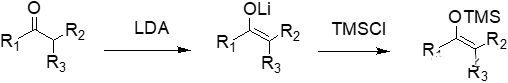
得到的外消旋混合物6不经过分离处理,直接加对甲苯磺酸(p-TSA),β-羟基脱水生成了α,β-不饱和酮7。中间体7的碳-碳双键用威尔金森催化剂((PPh3)3RhCl)和氢气进行催化氢化还原,得到中间体8。中间体8结构中的酮羰基再经过CBS对映选择性催化还原后得到带醇羟基的中间体9。
CBS还原反应即为科里-巴克什-柴田还原反应(Corey-Bakshi-Shibata(CBS)Reduction)。该反应所用的CBS催化剂是不对称还原反应中重要的手性催化剂,是利用脯氨酸为原料合成得到的手性硼杂恶唑烷硼烷催化剂,在硼烷存在下,可对酮进行不对称催化还原。不管是环状的酮还是链状酮,都可以被还原成高对应选择性的醇,其产物的手性构型和催化剂的构型有关,R-构型的CBS催化剂往往得到高选择性的S-构型的产物。研究还发现,CBS还原反应受到温度的影响,通过控制温度可以有效的控制反应进行和产物的对应选择性。
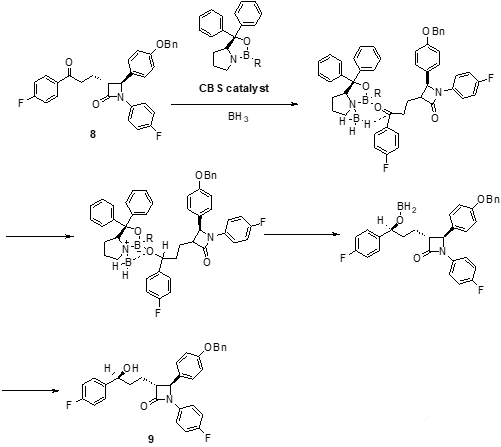
中间体9再经过钯碳(Pd/C)催化氢化之后脱去苄基(Bn)并最终得到依泽替米贝。
除了上述方法,中间体7也可以用Pd/C和氢气(H2)直接一锅法进行双键的加氢还原和苄基的脱除得到中间体10,中间体10再经过六甲基二硅脲(BSU)的处理,将结构中的酚羟基进行硅烷化保护,再进一步用CBS对映选择性还原结构中的酮得到醇羟基中间体12,12经过HCl处理后脱去三甲基硅基得到依泽替米贝。中间体10到依泽替米贝的三步合成可以通过一锅法高效实现。

除了上述两种合成方法之外,第三种合成方法路线如下:
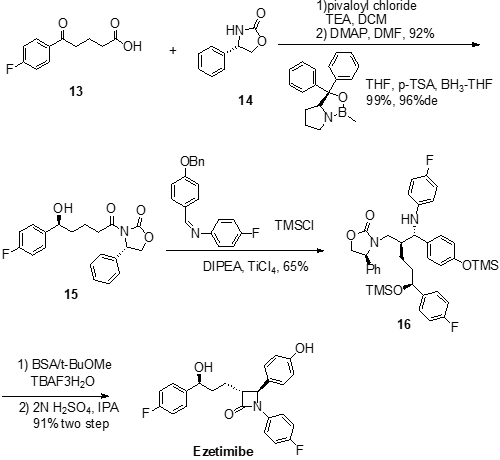
原料13首先和三甲基乙酰氯(pivaloyl chloride)反应,将羧基酰氯化,三乙胺(TEA)在起活化促进反应的作用。生成酰氯活性中间体后,进一步和手性辅助结构14中的胺基反应生成酮基酰胺中间体,反应中用4-二甲氨基吡啶(DMAP)催化促进反应进行。生成的产物进一步用CBS进行对映选择性催化还原,将酮羰基还原为醇羟基,得到中间体15,该反应过程选择了对甲苯磺酸(p-TSA)作为Lewis酸。以N,N-二异丙基乙胺(DIPEA)做碱,中间体15在四氯化钛(TiCl4)催化下与N-(4-苄氧基)甲苯-N-(4-氟)苯基亚胺发生亲电加成反应(类似于Michael加成反应),生成的中间体进一步用三甲基氯硅烷(TMSCl)处理,将活性羟基进行硅烷化保护得到中间体16。中间体16首先用牛血清蛋白(BSA)和四丁基氟化铵(TBAF)处理,水解后发生分子内成环,生成四元环内酰胺中间体,之后再用稀硫酸溶液处理,脱去分子结构中的三甲基硅基保护基(TMS)并最终以91%的两步总收率得到目标产物依泽替米贝。
Jin Li and Kevin K.-C. Liu Synthetic Approaches to the 2002 New Drugs.Mini-Reviewsin Medicinal Chemistry, 2004, 4, 207-233.