
手机扫码访问本站

微信咨询
烯丙基胺是天然产物,农用化学品和药物中普遍的结构基序,许多人致力于研究烯丙基胺的有效合成方法。现存在的主要方法有过渡金属催化的C-N键的合成;后过渡金属催化的二烯烃的区域选择性C-N键的生成。其中,炔烃也可以作为底物通过异构化得到丙二烯,然而这就限制了底物只能使用烷基炔烃。所以人们又发展了一种通过炔烃和胺的α-C位形成C-C键来得到烯丙基胺。
Buchwald课题组(图1)提出炔烃插入A中的Zr-C键得到一个五元环中间体B,水处理后得到烯丙基胺产物,和锆的氧化产物,然而五元环B的质子分解过程受到限制,使得这一反应无法进行催化循环。本文作者旨在找到一种锆催化剂可以降低质子分解过程的障碍,从而实现反应的催化循环。

图 1
先前,作者就报道过Zr(NMe2)4作为催化剂,用具有空间位阻的N-(三甲基硅烷基)胺进行烯烃的氢氨基烷基化。在加氢胺化催化中,发现了含有双(脲)配体的锆配合物容易形成七配位配合物,可用于有利于中性胺的配位,以实现所需的缔合催化转换步骤。作者认为这一催化剂参与的炔烃的氢氨基烷基化反应。
作者做了炔烃加氢氨基烷基化的化学计量制备的模型中间体。N-三甲基硅苄胺和甲苯与催化剂络合得到6配位的中间体1,然后在C6D6溶剂中与两当量吡啶反应脱去一当量的甲苯得到7配位的络合物2。在2的甲苯溶液中,加入一倍当量的二苯乙炔通过1H NMR检测到有五元金属环3生成。由于中性吡啶供体在Zr-C键旁配位,因此在配位仲胺存在下将引发3的质子分解。
为了探究这一质子分解过程,作者在3的氘代苯溶液中加入3.5倍当量的四氢吡咯,在室温下反应5 min后,通过1H NMR检测到3完全被消耗,生成了已知的络合物4,吡啶和烯丙基胺5a,该过程可以通过简单的胺底物而不是质子化学计量后处理来实现产品的轻松释放。

图 2
在此研究基础上,作者在最佳反应条件下使N-三甲基硅烷取代的胺底物与二苯乙炔反应,以60%的产率得到目标产物5a,但是有14%的副产物5b生成。而用N-苯基取代的胺底物参与反应,可以以82%的产率得到目标产物6a,且没有氢胺化的副产物6b产生。

图 3
接着,作者以N-苄基苯胺为模板底物对炔烃进行了拓展。当苯环的对位存在给电子或吸电子基团时,产率都有一定程度的下降,且没有区域选择性。而当其中一个苯基被2-吡啶基或三甲硅烷取代时,可以得到单一的异构体11或14,但是产率只有49%和31%。
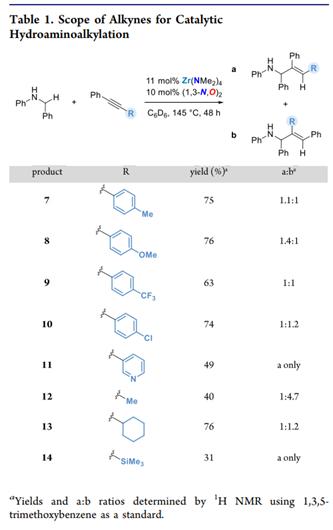
图 4
最后,作者以二苯基乙炔为模板底物对胺进行了拓展。当苯基的对位有甲基取代时,只需反应24h就可以以84%的产率得到目标产物15。而当有卤素原子取代时,产率都有所下降。然而当苄基的对位存在吸电子或给电子取代基时,对反应的影响都不大。

图 5
总之,本文实现了炔烃的催化加氢氨基烷基化以产生烯丙基胺产物。发现带有有利于形成七配位配合物的双(脲酸酯)配体的锆配合物可促进催化反应所需的化学计量转化。其中炔烃上的取代基对反应的区域选择性有较大的影响,胺的苯基上对位的取代基为给电子时,产率较高,取代基为吸电子时产率有所下降。
Zirconium-CatalyzedHydroaminoalkylation of Alkynes for the Synthesis of Allylic Amines
https://dx.doi.org/10.1021/jacs.0c10405