
手机扫码访问本站

微信咨询
药品名:地氯雷他定 Desloratadine
所属家族:抗组胺药物(第三代),三环类组织胺拮抗剂
主要应用:用于缓解慢性特发性荨麻疹及常年过敏性鼻炎的全身及局部症状
药效优点:无中枢神经系统和心脏毒性;可显著降低ROS含量和SOD活性即体内抗氧化活性(对慢性荨麻疹治疗更好)
组胺由组氨酸脱羧而形成,广泛存在于动植物体内。组织中的组胺以无活性的结合型存在于肥大细胞和嗜碱性粒细胞的颗粒中,以皮肤、支气管粘膜、肠粘膜和神经系统中含量较多。当机体受到某种刺激引发抗原-抗体反应时,引起肥大细胞的细胞膜通透性改变,释放出组胺,与组胺受体作用产生病理生理效应。抗组胺药物通常通过拮抗体内的组胺受体(H1 受体),从而阻断组胺与受体的结合来降低组胺对于人体的影响。
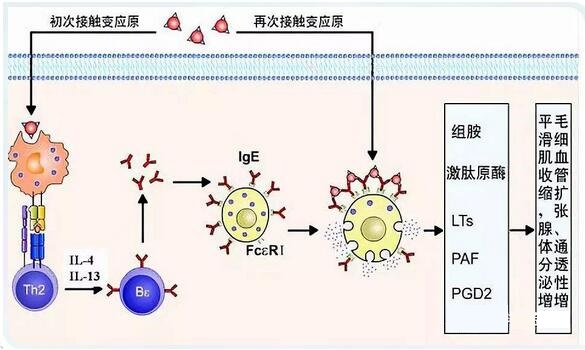
①依赖组胺受体的抗炎作用——地氯雷他定通过选择性拮抗 H1 受体,降低受体敏感性,阻断组胺刺激产生的多种炎症介质的释放
②不依赖受体的抗炎作用——
*抑制IgE 介导和非介导的肥大细胞和嗜碱性粒细胞组胺的释放
*抑制佛波酯 12-豆蔻酸 13-促分泌素诱导的肥大细胞释放 IL-13、IL-6、TNF-α 和 GM-CSF,
*抑制嗜酸性粒细胞的趋化黏附和浸润;
*抑制 NF-κB 活性,下调细胞因子和黏附分子的转录和表达,从而发挥抗炎效应!
③通过抑制 PERK1 /2 和 NF-κB 通路,从而抑制细胞因子如 IL-6、IL-8 和 GM-CSF 的生成(尚未证实PERK1/2通路活性)。
1565年莱昂纳多·博塔罗最早记录了枯草热病例(花粉使人打喷嚏、流泪和流鼻涕),并将其称为“玫瑰热”。
1902年过敏反应被提出并于1906年被定义。
1903年德国医生威廉·邓巴证明了枯草热以及这些应激反应是机体对花粉的反应产生的某种毒素释放所造成的。
1904年戴尔在研究黑麦的毒性时,发现了一种他命名为“组织胺”的物质
1910年戴尔和Patrick Laidlaw一起在《the journal of physiology》发表研究报告组胺的生理作用
1937年Daniel Bovet(意大利)和他的学生研究出了第一个抗组胺药(胸氧乙基二乙胺) 但因为抗组胺活性弱而毒性反应强未应用于临床。
1942年Bernard Halpern(法国)研制出第一个应用于人体的抗组胺药芬苯扎胺:
同年George Rieveschl(美国)发明第二个可以用于人体的抗组胺药苯海拉明
1944年Daniel Bovet发明马来酸拉明
从上世纪80年代开始,为改善和克服第一代 抗组胺药的缺点,新一代或称第二代抗组胺药先后问世,并逐渐替代经典的抗组胺药。
1981年9月,特非那定在经过许多科学家的研究和不断地结构修饰,由美国MerTeli公司开发上市
紧随其后,氯雷他定、阿司咪唑等药物接连被研发上市。
1967年Dr Frank J.iVllani首次合成4一氮杂一8氯一10,11一二氢二苯[a,d]环庚烯一5一酮
即三环酮(酸关环的方法),并发现此结构化合物具有抗组胺作用
1972年Dr Frank J.iVllani再次发现酰胺化合物直接关环制得三环酮
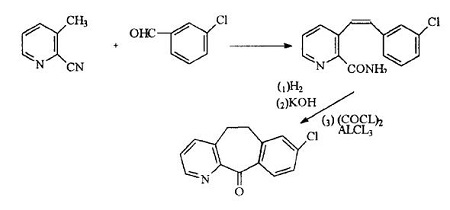
1981年Frank J. iVllani首次通过三环酮途径合成氯雷他定,并提出具有抗组胺性;
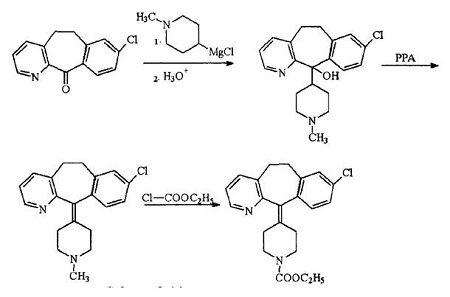
在经过长时间的结构确定和药理实验、临床实验后,由Schering-Plough公司开发的非镇静性长效,强效抗组胺剂,中枢神经系统副作用轻微的药物氯雷他定.1988年先后在比利时,菲律宾、加拿大美国和法国上市。
自此第二代组胺药物因 H1 受体选择性高, 无镇静作用,抗胆碱作用与抗组胺作用强而正式在过敏药物中崭露头角。
自 1986年以来,有关 NSA 类药物诱发的心脏事件和死亡危险性报告相继增多,引起了极大关注,WHO 药物不良反应协作中心于1986~1996年 共收到17个国家、976例抗组胺药的不良反应报告。诱发心脏毒性较多的是特非那定 ,其次是阿司咪唑, 氯雷他定和西替利嗪。
非索非那定
德国赫美罗药厂从1995年开始全面研究特非那定的活性代谢产物-非索非那定并获得了丰硕成果 ,作为新型抗组胺药于1997年经 FDA 批准问世。
地氯雷他定
氯雷他定研究完成后,其衍生物也不断开启研究,1988年
Dr Frank J. iVllani就提出了脱羧获得地氯雷他定DCL的路线并成功制得该药,经过药效评价和多次临床试验,于1998年在美国申请专利并上市。