
手机扫码访问本站

微信咨询
2,3-二氟苯乙酸是一种广谱使用的医药中间体,也是制备新型液晶的重要原材料,市场需求大,前景广阔。
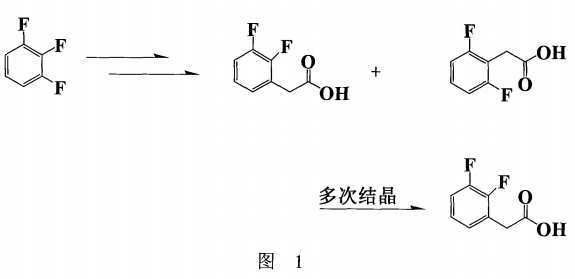
在2,3-二氟苯乙酸的合成策略中,由于其结构的特殊性,如拟在紧邻氟原子的碳上接上一个基团,用传统的富克反应法,所得产物的位置异构物多,不仅难以分离而且目标物并不是主产物;采用一般的亲核取代反应,如以1,2,3,-三氟苯为反应主体(图1),一者原料价格昂贵,再者副反应所产生的异构物不易除去(须结晶多次方可得纯的产品),造成收率偏低(<30%),不符工业化要求(B.A.Kowalczyk,Synthesis,1411(1997))。
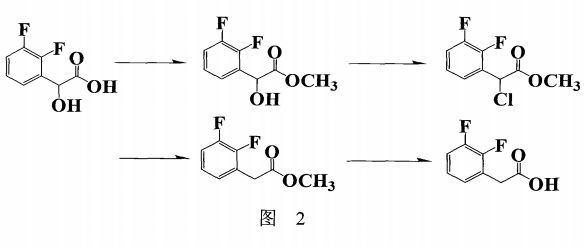
德国专利WO2008078350(图2)报导的以2,3-difluoro-mandelic aid为起始物经 酯化,氯化,还原,再碱解并酸化后制备2,3-二氟苯乙酸,由于步骤繁多,造成收率偏低(44.2%),而且起始物价格昂贵,也不适合工业化生产。
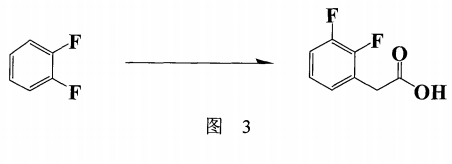
另外,日本专利:JP2004352724(也申请中国专利:CN100378062C)(图3)提出以邻二氟苯为起始物,在二种催化剂的存在下和正丁基锂及2-溴乙酸乙酯作用产生2,3- 二氟苯乙酸乙酯,收率最高为77%,其后再进行皂化反应及重结晶,则所得产物之总收率降至51.92%,再者其所使用的催化剂对其反应结果影响甚大(总收率8 %~77%),但此催化剂[二(乙酰丙酮-2,2’-联吡啶镍(II)],由于结构特殊不易买到,也不知是否可以重复套用以降低成本。如要自行制备此类有效之催化剂可能须耗费昂贵的代价,所以也不是付诸工业化最佳之途径。
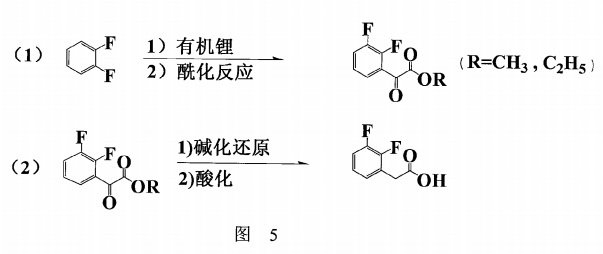
CN200910030821.9提供一种工艺流程简单,反应条件温和,后处理清洁简便且产物纯度高,成本低廉并适合工业化生产的制备方法。其具体步骤为:
(1)在低温下,邻二氟苯与有机锂作用生成邻二氟苯基锂,该邻二氟苯基锂再与草酸脂或氯草酸酯反应,生成2,3-二氟苯乙酮酸酯;
(2)将所述的2,3-二氟苯乙酮酸酯在碱性条件下进行还原,再经酸化析出得到2,3-二氟苯乙酸,然后结晶纯化。如图5所示:
具体操作如下:
在一个2L之三口瓶(A)中加入50g(0.438mol)之邻二氟苯及400ml之无水THF,然后密闭充N2,随后降温至-70℃并开始滴加176ml(0.440mol)之2.5M 正丁基锂(共用时2h),滴毕后于-65~-75℃保温继续搅拌2h;在此同时,于另 一个2L之三口瓶(B)中置入67.2g(0.46mol)之草酸二乙酯及100ml之无水 THF,然后密闭充N2并随之降温至-70℃。利用N2将瓶(A)之物料慢慢压入瓶(B)中(用时1h并控温在-65~-70 ℃区间内),之后于此温度范围内继续反应1h并自然开温至0℃(用时1h),随后滴加60ml,6N HCl于前述反应混合物中(用时30min,温度开至15℃,PH 3~4),滴加完HCl后,将此混合物继续搅拌30min并静置分层,将下层乳状液水层 以80ml之乙酸乙酯萃取二次,合并有机层后直接脱溶并蒸馏纯化,得2,3-二氟苯乙酮酸乙酯(淡黄色液体):85.1g,含量:96%(HPLC),收率:90.8%。
在一个1L单口瓶中置入91g(0.425mol)之2,3-二氟苯乙酮酸乙酯并加270ml H2O稀释,然后滴加57.3g(0.51mol)之50%KOH并继续搅拌1h(TLC显示已完全转化为钾塩);随后加入90ml正己烷并搅拌30min以去除有机层杂质,将 分出之水层移至另一干净之1L之三口瓶中,于室温(20~25℃)下加入180mlH2O 稀释,然后滴加57.3g(0.51mol)之50%KOH及39.9g(0.638mol)之80%水合肼 (各用时30min,温度~28℃),再继续搅拌反应1h,其后于30min内将反应温度升至85~95℃并继续回流反应16h。
随后将反应系统温度降至室温(用时1h)并滴加230ml之6NHCL和调节 PH至1(用时30min,温度~30℃)再继续搅拌30min,于此同时析出大量白色固体,经过滤并以100mlH2O洗2~3次直至滤出之水液PH 3~4,接着将白色固体抽于并放置烘箱(~85℃)烘于4h,得69.5g之2,3-二氟苯乙酸粗品,含量(HPLC)>96%。前述粗品以甲醇和H2O=1∶3结晶,得类白色2,3-二氟苯乙酸纯品62.53g,含量(HPLC):99.6%,收率85.5%。
[1] CN200910030821.9 2,3-二氟苯乙酸的工业制备方法